

CASO CLÍNICO
Niño de 4 años con manchas café con leche que han ido aumentando de tamaño y número desde el nacimiento. A la exploración presenta: Peso: 16.4 Kg (p34, 0.43 DE) Talla: 104.5 cm (p57, 0.19 DE) IMC: 15.02 (p25, 0.69 DE) Tensión arterial: Sistólica: 100 mmHg (p70, 0.55 DE) Diastólica: 48 mmHg (p42, 0.22 DE) Múltiples manchas café con leche de 1 a 3 centímetros de diámetro generalizadas (Fig. 1) no se evidencian otras alteraciones. Desarrollo neurológico sin alteraciones, acorde a edad según escala Denver 2.
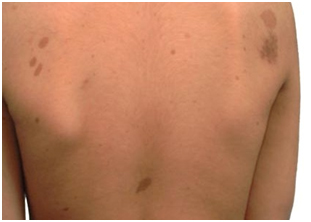
Figura 1 Manchas café con leche
Antecedentes |
Edad gestacional: 38s. Peso: 2390 gr. (p4, -1.88 DE) Madre, tía materna y abuela materna con manchas café con leche no estudiadas. Tía materna y abuela materna talla baja.
A los 9 meses de edad realizan resonancia magnética cerebral y medular normal.
Se solicita estudio genético que permite evidenciar mutación en heterocigosis p.Glu1549Valfs*, compatible con el diagnóstico de neurofibromatosis tipo I.
¿Cómo haría el seguimiento de este paciente? :
a) Control anual de resonancia magnética cerebral y medular.
b) Resonancia magnética solo en caso de sintomatología sugestiva de complicaciones neurológicas.
c) Control de presión arterial, visita anual con el oftalmólogo, valorar anomalías esqueléticas.
d) b y c son correctas.
En el año 2010 la Asociación española de Pediatría en su sección de protocolos en genética hace las recomendaciones para el seguimiento de los pacientes con neurofibromatosis tipo 1 señalando que no es necesario la realización rutinaria de estudios de imagen en tórax, abdomen y sistema nervioso a menos que el paciente tenga sintomatología sugestiva de complicaciones o se identifique una deleción completa del gen NF1 que se asocia a anomalías estructurales cerebrales. Así mismo señala que en los controles rutinarios de estos pacientes se les debe medir la presión arterial para descartar hipertensión, valorar la presencia de anomalías músculo esqueléticas como escoliosis y solicitar un control anual por el oftalmólogo (d) Respuesta correcta.
Neurofibromatosis tipo I (NF1)
Generalidades |
La NF1 es una enfermedad progresiva multisistémica autosómica dominante de expresividad variable y con un curso difícil de predecir. Afecta principalmente los sistemas dermatológico, neurológico, esquelético, oftalmológico y endocrino. Tiene una prevalencia de 1/3000 personas.
El gen se localiza en el brazo largo del cromosoma 17, codifica una proteína la neurofibromina que actúa como un supresor tumoral al inhibir la actividad de la proteína ras, la cual está implicada en el crecimiento y la proliferación celular, lo que explicaría la tendencia a la proliferación en individuos afectos de la enfermedad.
El 50% de los casos se originan por mutaciones de novo. En los niños que tienen historia familiar, el diagnóstico es relativamente sencillo, porque requieren solo un signo adicional al del padre afectado y éste por lo general está dado por las manchas café con leche.
Diagnóstico |
Desde 1.987 el Instituto Nacional de Salud de los Estados Unidos definió los criterios diagnósticos de la NF1, que establece que un paciente debe cumplir al menos 2 de los siguientes 7 criterios:
- Presencia de seis o más manchas café con leche iguales o mayores a 5mm en pacientes prepuberales y mayores de 15mm de diámetro en pacientes postpuberales.
- Existencia de 2 o más neurofibromas periféricos de cualquier tipo o bien un neurofibroma plexiforme.
- Presencia de pecas (efélides) en axilas, ingles y base del cuello (signo de Crowe)
- Glioma del nervio óptico.
- Presencia de dos o más nódulos de Lisch.
- Presencia de una alteración ósea definida como displasia del esfenoides que en los casos de mayor intensidad se acompaña de otras anomalías.
- Afectación de un familiar de primer grado (padre, hermano, hijo)
A los pacientes que cumplan los criterios de NF1 se les debe solicitar el estudio genético para confirmar el diagnóstico.
CLÍNICA |
Varía con la edad del paciente. A lo largo de los dos primeros años se suelen observar manchas café con leche (hallazgo más frecuente), macrocefalia, talla baja y diversas anomalías torácicas. En la edad preescolar se observan efélides en axilas, ingles y base del cuello. En la adolescencia pueden aparecer nódulos de Lisch y neurofibromas. En la edad adulta están presentes todos los síntomas mencionados.
Diagnóstico Diferencial |
Cuando el diagnóstico no está claro se deben considerar otras formas de neurofibromatosis, otras enfermedades con manchas café con leche, enfermedades con alteraciones de la pigmentación, síndromes de hipercrecimiento localizados y tumores que se confunden con neurofibromas.
- Dentro de las otras formas de neurofibromatosis, la que le sigue en frecuencia es el tipo 2: se manifiesta con hipoacusia que suele ser bilateral por la presencia de neuromas acústicos con compromiso de otros pares craneales, de las raíces nerviosas raquídeas, meningiomas y ependimomas, con escasas manchas café con leche.
- Dentro de otras enfermedades que cursan con manchas café con leche encontramos:
o Síndrome de McCune-Albright en el cual las manchas café con leche suelen ser de tamaño variable, de bordes irregulares, no suelen pasar la línea media, pueden tener distribución segmentaria y con frecuencia predominan en un lado del cuerpo. La enfermedad cursa con la tríada: manchas café con leche, displasia fibrosa del esqueleto y pubertad precoz.
o Esclerosis tuberosa: pueden coexistir manchas café con leche con lesiones acrómicas, angiomas o zonas de atrofia cutánea, asociado a evidentes signos neurológicos de retardo del desarrollo y epilepsia.
- Las enfermedades por alteraciones de la pigmentación suelen cursar con nevus hiperpigmentados los cuales pueden confundirse con manchas café con leche, por lo general se asociación a retardo del desarrollo, retardo mental, hidrocefalia o epilepsia.
Complicaciones |
El 50% presentan formas leves de la enfermedad y un tercio desarrollan formas graves en las cuales habrá afectación de los diferentes órganos y sistemas por los neurofibromas, variando su expresión clínica en función de la edad. Las principales complicaciones son: pseudoartrosis, escoliosis, trastornos cognitivos y del aprendizaje, convulsiones, tumores del tronco cerebral y cerebelo, los gliomas de la vía óptica, talla baja, alteraciones de la pubertad, los tumores espinales y paravertebrales, cefalea, esclerosis del acueducto con hidrocefalia secundaria, trastornos cerebro vasculares, meningocele, diversos tumores malignos de los nervios periféricos, feocromocitoma, somatostatinomas, hipertiroidismo, diabetes mellitus y déficit de hormona de crecimiento.
Seguimiento |
La finalidad del seguimiento de estos pacientes es diagnosticar tempranamente la aparición de complicaciones y mejorar la evolución de las mismas.
Al momento del diagnóstico en edad prepuberal se sugiere:
1. Evaluación oftalmológica completa: Fondo de ojo, campimetría, agudeza visual, visión de colores.
2. Evaluación neurológica: Evaluar algún déficit neurológico, monitoreo del desarrollo intelectual (usar escalas objetivas como por ejemplo la Escala Denver 2)
3. Evaluación antropométrica y presencia de caracteres sexuales secundarios.
4. Evaluar presencia de escoliosis: derivar a traumatología si fuera necesario.
5. Control dermatológico por especialista del área.
6. Medición de la presión arterial.
7. Evaluación cardiológica (descartar estenosis pulmonar)
8. Evaluación y asesoramiento genético.
9. Evaluación de los familiares de 1er. grado por dermatología y oftalmología.
Luego del diagnóstico se sugiere (en niños asintomáticos) hasta el inicio de la pubertad, un control anual de los puntos del 1 al 6, el control por el dermatólogo se realizará en cualquier momento en los que aparezcan lesiones de crecimiento rápido o sospecha de malignización.
No se recomiendan los estudios de imagen a menos que existan síntomas sugestivos de complicaciones o por la presencia de una deleción completa del gen NF1 que se asocia a anomalías estructurales cerebrales. Si la evaluación oftalmológica no es posible o confiable en niños menores de 4 años se debe realizar una resonancia magnética cerebral para visualizar la presencia o no de glioma del nervio óptico; así mismo si hay síntomas visuales, alteración del examen oftalmológico o neurológico, o signos de pubertad precoz está indicada la realización de una resonancia magnética cerebral.
Se debe siempre evaluar hipoacusia y problemas de audición, los cuales suelen ser progresivos y manifestarse como tinnitus, en caso de síntomas o dudas diagnosticas se debe referir a otorrinolaringología para que realice las pruebas de audición pertinentes.
Una vez que se hace el diagnóstico se debe ofrecer apoyo psicológico a la familia, además en la etapa escolar y adolescencia se debe explicar a los padres y a los pacientes problemas del entorno sexual y reproductivo.
Se debe dar consejo genético a los padres y a los pacientes cuando decidan serlo.
DIAGNÓSTICO FINAL |
Neurofibromatosis tipo I
Bibliografía |
Clementi M, Milani S, Mammi I, Boni S, Monciotti C y Tenconi R. Neurofibroatosis type I growth charts. Am J Med Genet. 1999;87(4):317-23.
Ferner RE, Huson SM, Thomas N, Moss C,Wills-haw H, Evans DG et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis I. J Med Genet. 2007; 44:81-8.
Guillén E, Ballesta MJ y Galán E. Protocolo de seguimiento de la neurofibromatosis tipo 1.
Asociación Española de Pediatría. Protoc diagn ter pediatr. 2010; 1:44-50.
National Institutes of Health (NIH) Consensus Development Conference Statement: Neurofibromatosis. Bethesda, Md., USA, July 13-15, 1987. Neurofibromatosis. 1988;1(3):172-8.
Yturriaga R. Neurofibromatosis tipo I (NF1) y pubertad. En: IV Symposium Ferring en Endocrinología Pediátrica. Madrid; Ferring Productos Farmacéuticos; 2008.p. 1-5












